

See the Latest Textbook on Orbital Tumors by Dr. BCK Patel MD, FRCS and Colleagues

The most current textbook on orbital tumors with detailed descriptions of conditions, diagnoses, differential diagnosis, best techniques of surgical and medical treatment and videos.
File Size: 136719 KB
Print Length: 327 pages
Publisher: Springer; 3 edition (June 17, 2019)
Publication Date: June 17, 2019
Sold by: Amazon Digital Services LLC
Language: English
ASIN: B07TDLNQMT
Text-to-Speech: Enabled
Enhanced Typesetting: Enabled
ISBN-13: 978-3030135577
ISBN-10: 3030135578
What is an orbital tumor
Is any tumor that occurs within the orbit of the eye. The orbit is a bony housing in the skull about 2 inches deep that provides protection to the entire eyeball except the front surface. It is lined by the orbital bones and contains the eyeball, its muscles, blood supply, nerve supply, and fat.
Tumors might develop in any of the tissues surrounding the eyeball and might also invade the orbit from the sinuses, brain, or nasal cavity, or it might metastasize (spread) from other areas of the body. Orbital tumors can affect adults and children. Fortunately, most are benign.


What causes orbital tumors?
Children
Most childhood orbital tumors are benign and are the result of developmental abnormalities.
Common orbital tumors in children are dermoids (cysts of the lining of the bone) and hemangiomas (blood vessel tumors).
Malignant tumors are unusual in children, but any rapidly growing mass should be cause for concern.
Rhabdomyosarcoma is the most common malignant tumor affecting children, and it usually occurs between the ages of 7 and 8.
Adults
The most common orbital tumors in adults are also blood vessel tumors, including hemangioma, lumphangioma, and arteriovenous malformation.
Tumors of the nerves, fat, and surrounding sinuses occur less often.
Lymphomas are the most commonly occurring malignant orbital tumors in adults.
Metastic tumors most commonly arise from the breast and prostate, while squamous and basal cell cancer can invade the orbit from surrounding skin and sinus cavities.
What are the symptoms of an orbital tumor?
Symptoms of an orbital tumor might include:
protrusion of the eyeball (proptosis)
pain
loss of vision
double vision
redness
swelling of the eyelids
obvious mass.
Prominence of the eyes is not necessarily the result of a tumor, but might result from inflammation such as that caused by Graves' thyroid disease.
In children, parents might initially notice a droopy eyelid or slight protrusion of the eye.
How are tumors diagnosed?
Orbital tumors are most commonly diagnosed with either a CAT scan or MRI. If either of those tests look suspicious, a biopsy might be performed.?
How are orbital tumors treated?
Treatment of orbital tumors varies depending on the size, location, and type.
Some orbital tumors require no treatment, while others are best treated medically or with the use of radiation therapy.
Som might need to be totally removed by either an orbital surgeon or a neurosurgeon, depending on the particular case.
After removal, additional radiation or chemotherapy might be required. Surgery has become much safer because CT scans and MRI testing can help pinpoint the location and size of the tumor.
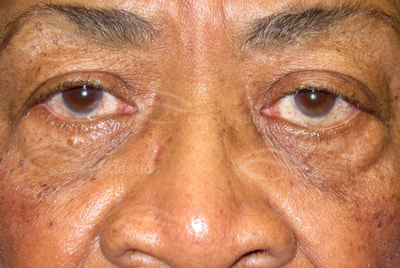
"Well, the last time I had a picture taken I could hardly see my eyes because of the weight of heavy eyelid. Then I paid attention to how I was actually using my eyes and I really noticed when I was looking at anything especially the computer I was straining my forehead to see better. Since I have had it done I no longer have to lift the forehead and tilt my head to see. It is amazing! I love..." D. Rock 63 Yrs Old with Fat Droopy Eyes - Salt Lake City, UT
Common Orbital Tumors in Adults
Introduction to Orbital Tumours
Gone are the days when every case of proptosis had to undergo a possibly futile biopsy procedure to the eyeball and face the consequences thereafter. The dawn of noninvasive radiodiagnostic techniques such as ultrasonography (USG), computerized tomography (CT) and magnetic resonance imaging (MRI) marked the end of this era of uncertainty. Biopsy is justified only in rare cases of malignancy or in lymphomatous lesions to confirm the histological variety.
About 20-25% of orbital disease are due to neoplasms and are commonly seen during and after the seventh decade of life. Malignant primary cancers of the orbit that call for a biopsy and radical surgery arise almost exclusively from the lacrimal gland.
Treatment Options
Signs and Symptoms
Orbital tumors are suspected by the presence of following signs and symptoms:
Proptosis or exophthalmos due to anterior displacement of the globe
Superior orbital tumours displace the globe inferiorly while masses in the inferior orbit displace the globe vertically.
Globe motility is limited by simple mass effect, extraocular muscle involvement, involvement of nerves to extraocular muscles, and adhesions of tumour to the globe.
Orbital pain, particularly in rapidly growing tumours.
Increased retropulsion, which means increased resistance to digital pressure applied on the side of the tumour.
Mass effect flattens the globe with changes in refractive power of the eye termed as a foreshortened globe.
Pressure on sensory nerves resulting in periocular paraesthesia or anaesthesia.
Proptosis causing droopy eye lid or ptosis.
Palpable mass, when tumour is anteriorly placed.
Visual field defect limited to affected eye only.
Obstruction of venous outflow leading to lid edema.
Papilledema results from mass effect on the retrobulbar optic nerve
Well-defined Orbital Masses: Well-circumscribed lesions of the orbit include cavernous hemangioma, neurofibromas, schwannoma, hemangiopericytoma, meningioma, and gliomas. Cavernous hemangioma is the most common benign orbital tumour in adults, similar to capillary hemangioma in children. Patients present with painless proptosis that is gradually progressive with features of mass indenting the globe, striae in the retina and flattened globe on imaging studies. Surgical treatment for en masse removal is advocated.
A mucocele or mucopyocele is the most common cause of proptosis in children, and presents as a cystic, encapsulated mass that usually originates from the nasal sinuses (usually frontal sinus), following repeated bouts of sinusitis. Surgical drainage is considered only after treatment failure with a course of antibiotics, or in the presence of optic nerve compression.
Diffuse Orbital Mass: These lesions usually require a biopsy to confirm the diagnosis. They include lymphoma, benign reactive lymphoid hyperplasia, orbital cellulitis, benign or malignant fibrous histiocytoma, neurofibromas, and sarcomas. Lymphomas are managed by radiation or chemotherapy, while pseudotumour is treated with steroids and ASA. Diffuse orbital masses requiring surgery include lymphangioma, fibrous histiocytoma, and neurofibroma.
Thyroid Ophthalmopathy: Many orbital tumours present with proptosis or bulging of the eyeball, the most common cause being thyroid-related immune orbitopathy. There may possibly or may possibly not be laboratory derangements in thyroid function tests. Thyroid ophthalmopathy is four to five times more common in women, particularly in their perimenopausal phase. History of cigarette smoking may possibly precede a more fulminant course. Pathogenesis of the disease is unclear leading to ambiguity in treatment protocols. Anterior segment eye complaints are treated with lubricants and topical steroids. Resultant glaucoma is managed with topical anti-glaucoma medications. Diplopia may possibly need steroids and surgery while proptosis might require steroids, radiation or corrective surgery.
Lacrimal Gland Tumours: They are easily diagnosed using ophthalmic B-scan at ultrasonography. More detailed analysis, particularly for bony involvement requires CT scans. Treatment comprises steroids, biopsy or excisional surgery.
Bilateral Masses are either inflammatory lesions such as sarcoid/pseudotumour, or lymphomas. Inflammatory lesions constitute an overwhelming majority and are usually tender.
Unilateral Masses are notorious and usually require a biopsy to rule out malignancy. Benign mixed tumours are painless, gradually progressive masses of the lacrimal gland causing inferior displacement of the globe among patients in their third and fourth decades of life. They are treated by surgical excision of the mass within its pseudocapsule.
Adenoid Cystic Carcinoma is the most common malignant epithelial tumour of the lacrimal gland and presents similar to a benign mixed tumour. However, there is pain, numbness, diplopia and visual disturbance and a circumscribed lacrimal gland mass with blurred margins infiltrating into bone on CT scan. Treatment is surgical. They may possibly recur years later but with a mortality of 90%.
Pleomorphic Adenocarcinomas are highly aggressive, malignant mixed tumours seen in patients in their fifth and sixth decades of life. It is the second most common malignant tumour of the lacrimal gland and presents as a painless mass with or without new increase in size. They usually have a history of prior biopsy for a benign mixed tumour which has undergone malignant transformation. Treatment is surgical but more than 75% of the patients die of metastases within 5 years.
Lymphoid Masses: Lymphomas require careful pathologic analysis aided by fresh tissue with appropriate marker studies and systemic staging, which help to define the disease and guide treatment with chemotherapy alone, local radiation alone, or a combination of both. Bilateral orbital involvement indicates a poorer prognosis.
Bone Lesions of the Orbit: CT scan provides the best delineation of bony lesions causing proptosis.
Primary Bony Lesions: Osteomas, osteogenic sarcoma, and fibrous dysplasia.
Secondary Bony Lesions: Metastases from prostate, thyroid, lung, breast, kidney, and the non-metastatic eosinophilic granuloma, otherwise known as histiocytosis X spectrum.
Cavernous Hemangioma
Orbital cavernous hemangioma is the among the most common benign neoplasm found within the adult orbit. It is a slow-growing, benign tumour involving vascular structures within the muscle cone of the orbit, which pushes the eyeball forward as it grows, resulting in proptosis. Bilateral cases are rare. It is most commonly reported in middle-age adults (20-40 years), with women more affected than men.
Symptoms of Cavernous Hemangioma
Cavernous hemangioma of the orbit presents with mass effect due to an increase in volume of the orbital contents. It causes the following symptoms and signs:
Painless, slowly progressive bulging of the globe
Decrease in visual acuity and visual field defects due to mass effect or involvement of the optic nerve, extraocular muscles or surrounding vasculature.
Double vision or diplopia due to extraocular muscle dysfunction or orbital axis mismatch between the two eyes.
Lagophthalmos due to extraocular muscle dysfunction or nerve involvement results in exposure keratopathy, keratitis, and corneal perforation.
Pupillary dysfunction due to involvement of neural structures within the orbit.
Diagnosis of Cavernous Hemangioma
A thorough ophthalmologic examination is key to formulating an exhaustive list of differential diagnoses.
A detailed history and review of symptoms is of paramount importance.
Examination should be thorough and should include observation and palpation of affected eye. Hertel exophthalmometry is done to document axial proptosis.
Assess near vision, distant vision, colour vision and visual fields followed by testing of pupillary and extraocular muscle function. Defect in any of these parameters signal compression of the optic nerve and imaging studies should be done. Extraocular muscle dysfunction is measured using prismatic evaluation.
Slit lamp or penlight evaluation might or might not detect any abnormalities.
Dilated fundoscopy might show choroidal folds due to mass effect while optic nerve compression shows up as visibleedema, elevation, pallor, or even atrophic changes in the fundus.
Cavernous hemangioma is suspected clinically and usually confirmed with orbital imaging studies. Following investigations help to evaluate the presence and extent of the disease:
CT scan detects an oval or round, homogenous mass with sharp margins, but falls short of a definitive diagnosis.
A-Scan Ultrasonography shows a uniform high-echogenicity while Doppler flow study reveals decreased blood flow within the lesion.
MRI of the cavernous hemangioma exhibits a homogenous signal. Gadolinium shows an initial central patch enhancement followed by total homogenous enhancement.
Imaging of Cavernous Hemangioma
CT: smooth discrete lesion, fills with dye after 20 min; coronal cuts important to know tumor position relative to optic nerve. for sugical plan
MRI: hypointense to fat on T1, hyperintense to fat on T2
U/S: high reflectivity (A-scan high amplitude internal echoes)
Treatment of Cavernous Hemangioma
Most cavernous hemangiomas require no treatment. The surgical approach, when indicated, depends on location and size of the tumour.
Cavernous hemangioma involving the anterior two-thirds of the orbit is resected via an anterior eyelid, transconjunctival or transcaruncular approach. A lateral orbitotomy or its variant is more appropriate for tumours located more posteriorly. Lesions involving the orbital apex warrants a transcranial approach.
A cryoprobe aids removal of well-circumscribed tumours with minimal risk of capsular rupture or blood loss. Carbon dioxide laser, Nd:YAG laser and Gamma knife surgery are newer modalities of treatment that can be considered.
The visual prognosis with complete excision is excellent but incompletely excised lesions are notorious for recurrences. Occasionally, visual loss can occur as a complication of surgery.




Pathology of Cavernous Hemangioma
well encapsulated and tolerated
shows large cavernous spaces with red blood cells
"I had a excellent eye lift done by Dr. Patel. He knows what he is doing and is very pleasant. Dr.Patel was easy to get an appointment and he works with you. The office staff was very pleasant and made you feel calm." D. Gull Highly recommended for eye lift surgery - Salt Lake City, UT
Orbital Tumors - Herniated Orbital Fat

orbital fat prolapse
Prolapse of subconjunctival intraconal orbital fat
First described in pathology literature in 2007 (Am J Surg Pathol 2007;31:193)
Clinical features
Rarely causes an intraorbital mass lesion
Mean age 66 years, 90% men
Prolapse is usually into superotemporal quadrant or lateral canthus
Usually due to orbital fat herniation through a dehiscence in Tenon's capsule
Manifests as unilateral or bilateral yellowish mass
Does not recur
Differential diagnosis
Pleomorphic lipoma: different clinical presentation; aggregates of bland spindle cells, floret cells and wiry collagen
Well differentiated liposarcoma: different clinical presentation; enlarged hyperchromatic cells within fibrous septae
Orbital Tumors - Hemangiopericytoma
Description of Orbital Tumors - Hemangiopericytoma
Contractile cells that wrap around the endothelial cells of blood vessels are known as pericytes. Orbital hemangiopericytoma is a rare, solid, slow-growing tumour that arises from the proliferation of pericytes in the orbital blood vessels but can involve blood vessels in the conjunctiva, choroid, optic nerve or skin of medial canthus.
Hemangiopericytoma constitutes 1-3% of all biopsied lesions of the orbit and 1% of all lacrimal sac tumours. This tumour might possibly be benign or malignant, and starts around 40 years of age with a predilection for men.
Pathophysiology of Orbital Tumors - Hemangiopericytoma
Orbital hemangiopericytoma is caused by inordinate layering of sheets of pericytes around improperly formed blood vessels within the orbital structures. The tumour cells contain few cytoplasmic organelles and are formed from pluripotent mesenchymal cells surrounding the blood vessels. “Staghorn” vessels are thin-walled, branching blood vessels seen specifically in hemangiopericytomas.
They have ovoid or spindle-shaped nuclei. 75% of the lesions are encapsulated and well-circumscribed while 30% of orbital hemangiopericytomas look very similar to a malignancy. They stain uniformly for CD34 and vimentin while 70% are positive for Leu-7.
Symptoms and Signs of Orbital Tumors - Hemangiopericytoma
Clinical presentation is variable. Patients might possibly present with any of the following symptoms:
Painful or painless, slow-growing mass in the orbit
Proptosis and exophthalmos
Raised pressure within the orbit
A painless mass near the medial canthus might possibly be a lacrimal sac hemangiopericytoma
Epiphora or watering of the eyes
Orbital hemangiopericytoma commonly involves the superior orbit and are commonly ballottable on palpation. There might possibly be visual loss, proptosis during exophthalmometry and decreased extraocular muscle motility depending on location of the tumour
Diagnosis of Orbital Tumors - Hemangiopericytoma
Even though proper clinical examination will aid in reaching a probable diagnosis, imaging studies like Computed Tomography or Magnetic Resonance Imaging help in planning for surgical excision. Definitive diagnosis is reached only through histopathologic evaluation of the tumour.
Hemangiopericytomas are easily confused with various orbital masses like fibrous histiocytoma, hemangioma, glomus tumour, sarcoma and vascular malformation.
Treatment of Orbital Tumors - Hemangiopericytoma

Definitive management of a case of hemangiopericytoma is complete local excision of the tumour along with the capsule. Maintaining proper haemostasis during surgery is of paramount importance as the tumour is highly vascular.
There has been some buzz about the role of chemotherapy and radiotherapy in preoperative management of the tumour, but with limited benefit.
There is an overall 89% 5-year-survival rate seen in hemangiopericytoma. There is also a possibility for local recurrence and local metastasis, but distant metastasis to lung, liver, bone and mediastinum is a rare occurrence.
Orbital Tumors - Lacrimal Gland Tumors
General
look for fullness of upper lid, asymmetry of superior sulcus, abnormal lid contour
majority lacrimal gland masses are idiopathic inflammatory dacryoadenitis
especially S-shape, often palpable
check for mobility, smooth, rubbery or nodular
proptosis is evidence of posterior growth, otherwise globe is down and media
Imaging
CT very good for differentiating inflammation from tumor: inflammation and lymphoid with in gland cause diffuse enlargement, elongated shape, contour around globe; neoplasms are isolated, globular, displace & indent globe



Pathology
Epithelial Tumors
50% benign mixed
Benign Mixed Tumor (Pleomorphic Adenoma)
most common epithelial tumor
30-50 year old
palpable, painless, slow (history often reveals symptoms > 1 year) growing with globe dispalced down, medial, axial proptosis
incites bony cortication, enlargement/expansion lacrimal gland fossa, firm lobular mass
lacrimal gland is oblong if inflammatory, globular if malignant
pathology
metaplasia of epithelial cells to form stroma, cartilage
benign epithelial cell nests with loose mesenchymal connective tissue
variability of above is mixed tumor
microscopic extension into pseudocapsule causes recurrence if margins not adequate at excision
treatment
must excise it all with lateral orbitotomy with en bloc excision including pseudocapsule
don&/260-Lacrimal Gland Tumors/#146;t biopsy b/c of 1/3 chance of recurrence, significant risk of malignant degeneration
50% carcinomas (50% of these are adenoid cystic, remainder: malignant mixed, 1o adeno carcinoma, mucoepidermoid carcinoma, squamous carcinoma)
Malignant Mixed Tumor
often arise from 1o benign mixed or from recurrent benign mixed if incomplete excision
path
similar to benign mixed but with malignant change
least common epithelial tumor
treatment
frequent exenteration, bone removal necessary
fatality rate of 50%
Adenoid Cystic Carcinoma (Cylindroma) (25% if epithelial lacrimal gland tumors)
most common (highly) malignant tumor of lac gland
PAIN from bone destruction, perineural invasion, rapid course differentiates from benign mixed
pathology
swiss cheese appearance, stain with mucicarmine, looks benign, infiltration of orbital tissue, incl. perineural invasion
basaloid pattern worst prognosis
treatment
radical orbital exenteration (of roof, lateral wall, floor, orbital soft tissue, anterior temporalis muscle), with XRT
death from intracranial extension or systemic metastisis after multiple recurrences
Non-Epithelial Lacrimal Gland Tumors
Inflammatory
1/2 of lacrimal tumors
Orbital inflammatory syndrome
Also used to be called pseudotumour
Sarcoidosis
Orbital Tumors - Lacrimal Sac
Lacrimal Sac Tumors
rare
mass above medial canthal tendon
complaints of epiphora or chronic dacryocystitis
irrigation could pass to nose or blood could reflux from punctum
CT could show extent of mass
DCG could show filling defect
could have skin ulceration, telangiectasia, + lymph nodes
could originate from skin or nasal mucosal tumors
benign squamous papillomas most common 1o
squamous cell carcinoma more than adenocarcinoma most common malignant tumor


Orbital Tumors - Lymphoma
General
almost exclusively in adults
continuum including benign reactive lymphoid hyperplasia (pseudolymphoma) to atypical lymphoid hyperplasia to low-grade then high grade malignant lymphoma;
also Orbital inflammatory syndrome pseudotumorplasmacytoma (including myeloma)
bimodal peak 30’s and 60’s
unilateral or bilateral
palpable rubbery mass fixed to orbital rim
maligant lymphoma & reactive lymphoid hyperplasia cause gradual (over a year or more) progressive, painless proptosis (vs. Obital inflammatory syndrome), lacrimal enlargement
usually on conjunctiva, anterior orbit so palpable or visible
eyelid or bilateral orbital involvement suggests systemic disease
putty-like molding to undisplaced tissues so little Visual Acuity (VA) or EOM loss; usually no bone erosion or infiltration unless high-grade lymphoma
lymphoma in retrobubar fat is infiltrative
Imaging
all patients w/ orbital lymphoid lesions need exam for systemic lymphoma (by oncology) with orbital, abdominal, chestCT;
CBC
bone marrow biopsy
chest x-ray (CXR)
bone/liver/spleen scan
Pathology
cytologic factors are more prognostic than mono/polyclonal; but
most benign lesions (reactive hyperplasia) are usually mostly T cells with polyclonal Bs;
malignant lymphoma usually more monoclonal B cells
both polyclonal and monoclonal varieties can develop systemic disease
open biopsy for path to give fresh tissue for touch preps; immunohistochemistry; flow cytometry; and gene rearrangement studies; in formalin for micro; gluteraldehyde for electron microscopy
Treatment & Course
X-Ray Therapy (XRT) for most orbital lymphoid lesions that are confined to orbit (50% of lymphomas)
Chemotherapy for systemic, therapy can be controversial
Course
up to 25% of patients have systemic lymphoma later on with benign reactive hyperplasia:
40% of patients get systemic involvlement within 5 years with atypical lymphoid hyperplasia:
Orbital Tumors - Neurofibroma
Description of Orbital Neurofibroma
Orbital neurofibroma is a peripheral nerve sheath neoplasm derived from Schwann cells, perineural cells and fibroblasts, and is probably the most common peripheral nerve tumour of the orbit. It constitutes about 0.8 to 3.0% of all histopathologically-proven lesions of the orbit. Orbital neurofibroma produce symptoms within the orbit, and may possibly or may possibly not be associated with systemic neurofibromatosis.
Orbital neurofibroma is classified into three subsets:
Plexiform neurofibroma is pathognomonic of neurofibromatosis.
Diffuse nerofibroma has variable association with neurofibromatosis.
Localized neurofibroma is rarely associated with neurofibromatosis.
Symptoms and Signs of Orbital Neurofibroma
The presence of multiple painful, well-circumscribed orbital tumours in a patient should raise the suspicion of neurofibroma. Typically, it presents with progressive symptoms of an orbital mass, including proptosis, globe displacement, impaired extraocular motility, ptosis, numbness, and rarely with decreased visual acuity. There may possibly also be associated features of neurofibromatosis. The clinical features of orbital neurofibroma is greatly dependent on its type and associated neurofibromatosis.
Plexiform orbital neurofibroma: The patient is usually a child in the initially decade of life with undeniable signs of neurofibromatosis, with about 66% having eyelid involvement. It usually begins as an eyelid mass that is more localized to its lateral third, giving the eyelid an S-shaped appearance. It may possibly extend further into the orbit, causing proptosis.
Diffuse orbital neurofibroma: The patient profile is similar to that of plexiform neurofibroma and usually presents with unilateral proptosis that may possibly or may possibly not involve the eyelids. It’s association with neurofibromatosis is not as strong as in plexiform neurofibroma.
Localized orbital neurofibroma: The typical patient is a young or middle-aged adult unlike the other forms which present in childhood. Clinical features include a solitary, well-circumscribed soft tissue tumour in the orbit causing proptosis and downward displacement of the globe. Less often, it can occur in the lacrimal gland and extraocular muscle, or even cause bony destruction to invade an adjacent sinus.
Diagnosis of Orbital Neurofibroma
Imaging studies are central to the evaluation of a suspected orbital neurofibroma. Both CT and MRI show smoothly marginated ovoid lesions, with or without lobulations.
Computerized Tomography: Shows lesions isodense or hypodense to extraocular muscles and shows variable contrast enhancement, with occasional ring-enhancement.
Magnetic Resonance Imaging: Demonstrates low-moderate T1 signal intensity and moderate- high T2 signal intensity. Heterogeneity of signal strength within the lesion is typical reflecting the mixed histopathology and vascularity of the tumours. Contrast enhancement is again variable.
Histopathology of a biopsy specimen or excised tumour confirms the diagnosis of orbital neurofibroma.
Treatment of Orbital Neurofibroma
The management of localized orbital neurofibroma consists of total excision, which is possible in about 46% of cases of isolated orbital neurofibroma. It also has the distinction of low recurrence after surgical excision. However, 72% of postoperative patients reported a sensory skin deficit.
However, the management of plexiform and diffuse orbital neurofibroma is complex, with an unpredictable outcome. Eyelid sparing orbital exenteration and orbital reconstruction is possibly the best treatment approach in patients with total eyelid ptosis and severe visual loss. Surgery is difficult due to diffuse infiltration and intracranial involvement. Recurrence after incomplete surgical removal is a common phenomenon. Hence, close follow-up is must.
Orbital Tumors - Schwannomas
Description of Orbital Schwannomas
Schwannomas are slow-growing, benign tumours that develop within the outer covering of peripheral and sympathetic nerves formed by Schwann cells, called the nerve sheath. They are sometimes referred to as neurilemmomas and commonly involve sensory and motor nerves supplying the orbital region. Schwannomas are rare tumours and constitute about 1% of all orbital tumours and 35% of peripheral nerve tumours within the orbit. It has a
It primarily affects individuals between 20 and 60 years old, with 10-15% cases accompanied by neurofibromatosis.
Pathophysiology of Orbital Schwannomas
Orbital schwannomas are seen as encapsulated growths within a peripheral or sympathetic nerve, which distinguishes them from a neurofibroma that affect nerve fibres themselves. They are benign tumours and are rarely associated with malignant transformation. They usually occur in the superior temporal region or muscle cone of the orbit pushing the eyeball forward and downward.
The orbital schwannomas has the following histologic components that help in a pathologic diagnosis:
Antoni A type areas: Constitute solid areas of tumour cells forming the bulk of the tumour.
Antoni B type areas: Contains loose cystic spaces with no axons.
Verocay body: Represents palisading of the tumour nuclei in acellular zones of the tumour.
Verocay bodies, when present, are a useful marker for orbital schwannoma. Histochemical staining with S-100 may possibly be positive.
Symptoms and Signs of Orbital Schwannomas
A patient with orbital schwannoma usually presents with a slowly progressive painless bulging of the eyeball, proptosis and takes years or even decades to produce symptoms.
Other symptoms and signs commonly seen are:
Oedema of the eyelids
Dystopia of the eyeball
Impaired ocular motility
Disturbances in vision including visual loss
Changes in the optic disc such as choroidal striae with hyperopia
Diagnosis of Orbital Schwannomas
Clinical history of a slow-growing tumour within the orbit producing symptoms hinting to a diagnosis of orbital schwannoma. This suspicion is further supported by the presence of an encapsulated mass.
Diagnosis is confirmed by Computed Tomography and Magnetic Resonance Imaging of the orbital region. Histologic diagnosis is obtained after surgical excision of the tumour and shows Antoni A or Antoni B areas, Verocay bodies and positive for S-100 immunohistological stain.
Treatment of Orbital Schwannomas
Complete surgical excision is the treatment of choice for orbital schwannoma. Being well-encapsulated, it is easily removed. There is however a small risk of recurrence. The risk of malignant transformation is minimal.
Orbital Tumors - Sphenoid Wing Meningioma
Go here to read the detailed article on sphenoidal wing meningiomas written by Dr. BCK Patel MD
Meningioma
A meningioma is a benign brain tumor. It originates from the dura mater, the tissue enwrapping the brain and spinal cord. Meningiomas are much more common in females, and are more common after 50 years of age. Of all cranial meningiomas, about 20% of them are in the sphenoid wing. In some cases, deletions involving chromosome 22 are involved.
Diagnosis of Orbital Meningioma
Sphenoid wing meningiomas are diagnosed by the combination of suggestive symptoms from the history and physical and neuroimaging by magnetic resonance imaging (MRI) or computer averaged tomography (CT). Tumors growing in the inner wing (clinoidal) most often cause direct damage to the optic nerve leading especially to a decrease in visual acuity, progressive loss of color vision, defects in the field of vision (especially cecocentral), and an afferent pupillary defect.
If the tumor continues to grow and push on the optic nerve, all vision will be lost in that eye as the nerve atrophies.Proptosis, or anterior displacement of the eye, and palpebral swelling may possibly also occur when the tumor impinges on the cavernous sinus by blocking venous return and leading to congestion. Damage to cranial nerves in the cavernous sinus leads to diplopia. Cranial nerve VI is often the originally affected, leading to diplopia with lateral gaze. If cranial nerve V-1 is damaged, the patient will have pain and altered sensation over the front and top of the head. Horner’s syndrome may possibly occur if nearby sympathetic fibers are involved.
Classification of Orbital Meningioma
Tumors found in the external third of the sphenoid are of two types: en-plaque and globoid meningiomas. En plaque meningiomas characteristically lead to slowly increasing proptosis with the eye angled downward. Much of this is due to reactive orbital hyperostosis.
With invasion of the tumor into the orbit, diplopia is common. Patients with globoid meningiomas often present only with signs of increased intracranial pressure. This leads to various other symptoms including headache and a swollen optic disc.
The differential diagnosis for sphenoid wing meningioma includes other types of tumors such as optic nerve sheathe meningioma, cranial osteosarcoma, metastases, and also sarcoidosis. Following the physical exam, the diagnosis is confirmed with neuro-imaging. Either a head CT or MRI with contrast such as gadolinium is useful, as meningiomas often show homogenous enhancement. Angiography looking for signs like stretched arteries may possibly be used to supplement evaluation of vascular involvement and to determine whether embolization would be helpful if surgery is being considered.
Treatment of Orbital Meningioma
Meningiomas have been divided into three types based on their patterns of growth. Histological factors that increase the grade include a high number of mitotic figures, necrosis and local invasion.
Treatment of sphenoid wing meningiomas often depends on the location and size of the tumor. Gamma knife radiationand microscopic surgery are common options. Their encapsulated, slow growth makes meningomas good targets for radiosurgery. In one series, less than one-third of clinoidal meningiomas could be completely resected without unacceptable risk of damaging of blood vessels (especially the carotid artery) or cranial nerves, risks that are lower with radiosurgery.
If surgery is done and the entire tumor cannot be removed, then external beam radiation helps reduce recurrence of the growth. Fortunately, most all meningiomas grow very slowly and almost never metastasize to other parts of the body. In part because of its slow growth, if a tumor is asymptomatic and found only by imaging, the best course is often observation with serial clinical exams and imaging. Possible indications for intervention would be a rapid increase in growth or involvement of cranial nerves. Untreated, one small series showed survival rates ranging from 5 to over 20 years, though most suffered unilateral blindness as well as paresis of extraocular movements.


Orbital Tumors Capillary Hemangioma
(Benign Hemangioendothelioma)
A capillary hemangioma (may also be called an "Infantile hemangioma," "Strawberry hemangioma", and "Strawberry nevus") appears as a raised, red, lumpy area of flesh anywhere on the body, though 83% occur on the head or neck area. These marks occur in about 10% of all births, and usually appear between one and four weeks after birth. It could grow rapidly, before stopping and slowly fading. Some are gone by the age of 2 , about 60% by 5 years, and 90–95% by 9 years.
Capillary hemangiomas occur 5 times more often in female infants than in males, and mostly in Caucasian populations.] Additionally, low birthweight infants have a 26% chance of developing a hemangioma.
It is the most common tumor of orbit and periorbital areas in childhood. While this birthmark could be alarming in appearance, physicians generally counsel that it be left to disappear on its own, unless it is in the way of vision or blocking the nostrils.


General
Color: strawberry or reddish-bluish-colored nevi
Texture: spongy
Location: upper eyelid more than lower eyelid; also occurs in the deep orbit (leads to proptosis)
Number: usually unilateral, often multiple
Family history: often
Size / Position: can increase in size with crying or positions in which they are lower to the ground
Presentation / Onset: appear in the initially eight l weeks of life
Rate of growth: rapidly for approximately six months to one year
Natural history: spontaneous involution starts approximately at one year through age 6
Incidence: occurs in approximately 1-4% of infants, could be more common in boys (3:2), more common in low birth weight infants.


Systemic Evaluation
occasional additional hemangiomas
usually no other significant involvement (EXCEPTION: Kasabach-merritt Syndrome)
Risks / Significance
Vision can be affected in several ways
inducement of strabismus which my lead to amblyopia
occlusion of the visual axis
inducement of astigmatism (related to position of tumor) and or myopia which could lead to amblyopia; refractive error could persist even after resolution of tumor
Differential diagnosisImaging

orbital cellulitis
rhabodmyosarcoma
lymphangioma
orbital dermoid
CT-well circumscribed lesion
MRI - well circumscribed lesion
Pathogenesis
poorly understood
Pathology
tumor composed of numerous closely packed capillaries.
proliferation of well-differentiated capillary endothelial cells
Treatment / Medical /Surgery
Not all orbital hemangiomas need to be removed. If, however, there is evidences of amblyopia or significant Ptosis ('Ptosisis may also be called Blepharoptosis. It refers to an eyelid which is droopy. This could cause a loss of vision, especially while reading, headaches, and eyebrow strain.') treatment could be initiated as outlined below:
Amblyopia management
Corticosterioid
Oral
Topical
Local injection
mixture of Kenalog and Celestone soluspan
Observation Radiation appropriate for small, non-visually threatening tumors
Interferon
CO 2 Laser
Orbital Tumors - Dermoids


Etiology
benign cystic lesions that are choristomas (tumors composed of tissues not usually found at the involved site)
originate at bony suture sites during embryogenesis as a result of surface skin elements becoming entrapped
usually found in early childhood (25% are noted at birth), but can also be diagnosed in adults
in adults, they often involve the deep orbital tissues and grow to a very large size
Not to be confused with Epidermoid cysts which are similar but lack certain elements in the wall of the cyst
Differential Diagnosis
encephalocele
lacrimal mass/tumor
lacrimal mass or tumor
Work-up / Course / Prognosis
CT/MRI imaging reveals characteristic round to oval shaped cystic lesion with a defined lining
Symptoms
painless
slow-growing
signs
typically do not displace the globe
typically do not elevate intraocular pressure
locations
occur in the superotemporal orbit (the most common site, 70% )
the superomedial orbit
deep orbit
Trauma might possibly lead to leakage of the cystic contents and likely will result in acute inflammation
Pathology
Lining
the cysts are lined by keratinizing, stratified squamous epithelium (85% of lesions), or
nonkeratinized stratified squamous epithelium
Filling
filled with keratin
hair shafts are usually present in lumen or wall of cyst (99%)
sebaceous glands are usually present in lumen or wall of cyst (75%)
sweat glands are often present in lumen or wall of cyst (20%)
Treatment
surgical excision is the treatment of choice. With complete excision, the prognosis is excellent.
usually once they child has reached age 12 months

Orbital Tumors - Lymphangioma

Description of Orbital Lymphangioma
Hemangioma and venous lymphatic malformation are the two most common orbital vascular lesions seen in the pediatric patient. Orbital Lymphangioma are benign hamartomatous tumours and could have an aggressive nature. Lymphangioma is rare, and constitutes less than 7% of orbital tumours. It usually presents with slow growth and a gradually progressive proptosis that is mild initially. The tumour is thought to be congenital, slow growing, and could not become clinically apparent for months or even years. It occurs in children and teenagers, but most commonly in the initially decade of life.
Symptoms of Orbital Lymphangioma
Most orbital lymphangiomas are initially asymptomatic, and progress gradually, manifesting as proptosis, eye pain and disturbed vision. Other symptoms include blurry vision, double vision, red eye, bulging eye, droopy eyelid, glaucoma, and vision loss. More than 50% of lymphangiomas affect anterior structures such as conjunctiva and adnexa. Orbital lymphangiomas are differentiated from hemangiomas in that orbital lymphangiomas never regress, while hemangiomas do. Spontaneous bleeding occurs within the tumour in 55% of patients causing cysts of blood called ‘Chocolate Cysts’. If the cyst forms behind the eye ball it causes proptosis, compressive optic neuropathy and loss of vision. Vascular changes can occur in the conjunctiva manifesting as “lympangiectasias”.
Diagnosis of Orbital Lymphangioma
Lymphangioma usually presents as a sudden painful bulging of the affected eye, with or without history of facial trauma or that the tumour or proptosis started right after an upper respiratory tract infection. Examination of anterior segment of the eye reveals bluish discoloration of blood vessels within the skin of the eye lid. The blood vessels can often extend underneath the conjunctiva, called lymphangiectasias. cases could be associated with corneal exposure, ulceration and optic nerve damage.
Ultrasound is the initially line of investigation for any patient with a typical history of pain with proptosis. To establish the diagnosis of lymphangioma, computed tomography (CT) or Magnetic Resonance Imaging (MRI) can be done, which can also demonstrate extra-orbital extensions.

Treatment of Orbital Lymphangioma
Orbital lymphangioma is an unusually slow growing tumour and commonly monitored by observation for growth, both clinically and radiographically prior to considering definitive intervention. The following flag signs are indicative of a more severe form of orbital lymphangioma and the need for active intervention:
Sudden spurt in tumour growth
Optic nerve compression
Corneal exposure problems like keratitis sicca
Glaucoma
Vision loss
Most orbital lymphangiomas are poorly defined in their extensions and hence en mass removal is out of the question. Most patients are treated with several debulking surgeries to relieve acute optic nerve compression or corneal exposure. In rare cases, these patients could require exenteration of the orbit, or radiation therapy for pain relief.
With regard to treatment, orbital lymphangioma is a challenging disease and often difficult to treat with good visual outcome. It is also notorious for cosmetic complications and the possibility of frequent recurrences.
Orbital Tumors - Optic Nerve Glioma
Clinical Symptoms
decreased visual acuity
minimal proptosis
restricted, decreased eye movement
strabismus
Etiology
neoplasm of the optic nerve
may also be called juvenile pilocytic astrocytoma
Optic nerve glioma in adults are glioblastoma
Optic nerve glioma is the most common cause of optic nerve enlargement
it accounts for 80% of optic nerve tumors
1% of all intracranial tumors
2% of childhood intraorbital masses
Demographics
80% are in children under 10 years old
peak age is 5-8
90% of patients are under the age of 20
more common in females
10-50% are in neurofibromatosis patients, especially if bilateral
15% of NF patients have an optic glioma
Clinical Course
Optic nerve gliomas grow very slowly and have similar pathology as juvenile pilocytic astrocytomas of the cerebellum.
typically intraorbitally and grow in a fusiform shape
25% are limited to the orbit
malignant degeneration is very rare in children
if the tumor begins in the chiasm, it is likely to invade surrounding parenchyma regardless of age
Imaging
On MRI, there is usually mild contrast enhancement. The affected optic nerve should be greater than 3 mm in diameter, or 1 mm wider than the unaffected side. It should be hypo- to isointense to muscle on T1 and hyperintense on T2. Fat supression sequences should be performed to see the entire extent of the lesion since it could appear to be more extensive than it is on T2-weighted images secondary to edema
Differential Diagnosis
Sarcoidosis
Infiltration by leukemia or lymphoma
Optic neuritis
Perineural hematoma
Papilledema of intracranial hypertension
Patulous subarachnoid space
Spindle Cell Sarcoma
Introduction to Spindle Cell Sarcoma Orbit
Spindle cell tumour is a type of connective tissue cancer which is characterized by the presence of spindle-shaped cells when seen under the microscope. It commonly occurs in the pleura, lungs, peritoneum, pericardium, liver, kidneys and rarely in the head and neck region.
Solitary fibrous tumour is a visceral spindle cell tumour originating from the mesenchyme and is rarely seen in the orbit. Orbital solitary fibrous tumours (SFT) remains a highly underdiagnosed tumour due to its rare occurrence outside of the pleural sites and its similarity with other spindle cell tumours.
Pathophysiology of Spindle Cell Sarcoma Orbit
Orbital solitary fibrous tumours show mesenchymal and/or fibroblastic differentiation and have a strong and diffuse immunoreactivity to CD 34. They are most commonly seen originating from the pleura and are rarely known to affect extrapleural sites like the orbit.
They are well-circumscribed, encapsulated tumours with cellular and cystic areas. The tumour cells have a characteristic spindle-shaped appearance and are arranged in whorls. Orbital solitary fibrous tumours stain positive for CD 34 and BCL-2 on immunohistochemistry.
Symptoms and Signs of Spindle Cell Sarcoma Orbit
Spindle cell sarcoma of the orbit is a slow-growing, mostly benign tumour presenting with gradual unilateral painless progressive proptosis and pressure symptoms in a middle-aged individual.
It becomes symptomatic after one month to several years after onset and may possibly be associated with visual loss, restriction of eyeball movement, palpable mass or blepharoptosis. The growth of orbital solitary fibrous tumour is usually variable, non-tender, and non-pulsatile.
Diagnosis of Spindle Cell Sarcoma Orbit
Solitary fibrous tumours of the orbit are usually diagnosed using various imaging modalities such as ultrasonography, Computerized Tomography, Magnetic Resonance Imaging, and Magnetic Resonance Angiography. Low reflectivity with moderated sound attenuation on ultrasonography might indicate the presence of a solitary fibrous tumour.
CT scan shows well-circumscribed mass with or without bony remodelling with moderate enhancement. MRI reveals intermediate to moderate intensity mass on T1-weighted images before and after gadolinium while angiography shows a richly vascular tumour with uniform staining.
Diagnosis is confirmed using histopathologic features and immunohistochemical studies.
Treatment of Spindle Cell Sarcoma Orbit
En bloc surgical excision with complete removal of the tumour is the treatment of choice for spindle cell sarcoma of the orbit.
The chance of recurrence leaves room for close clinical follow-up and review surgery, but there is no conclusive evidence supporting radiotherapy or chemotherapy for prevention of recurrence.
Fibrous Dysplasia
Description of Fibrous Dysplasia
Fibrous Dysplasia is a benign, slowly progressive disease of the bone, and usually presents in children and young adolescents. In this disease, normal cancellous bone is replaced by fibrous tissue and immature woven bone. It was originally described by von Recklinghausen in 1891, and given the name ‘fibrous dysplasia’ by Lichtenstein in 1938. The etiology of fibrous dysplasia is usually unknown.
There are three forms of the disease: 70% cases are ‘monostotic’ involving only one bone or contiguous bones, while 30% of cases are ‘polyostotic’ involving several distinct areas of the skeleton, and Polyostotic fibrous dysplasia as a part of McCune Albright Syndrome, accompanied by skin pigmentation and endocrine disorders constituting 3% of polyostotic cases.


Symptoms and Signs of Fibrous Dysplasia
Fibrous dysplasia most often presents with pain, swelling and disfigurement due to fibroblastic expansion of the involved parts. They also present with visual impairment, bulging of the eyeball, craniofacial swelling, headaches, periorbital pain, and epiphora.
The most common neurologic complications of fibrous dysplasia are visual impairment and hearing loss. Proptosis is the most common sign, with downward displacement of the eyeball and loss of visual acuity close at its heels. Other signs include sinus collapse, nasal obstruction, extraocular muscle palsies, trigeminal neuralgia, anomia and epiphora.
Visual impairment and hearing loss constitute the most common neurologic complications of fibrous dysplasia. About 0.5 - 1% of all cases of fibrous dysplasia go on to turn malignant, the most common malignancy being osteogenic sarcoma, followed by fibrosarcoma, chondrosarcoma, and malignant fibrous histiocytoma.
One should always check for café-au-lait spots in patients with fibrous dysplasia as it might be part of McCune-Albright syndrome.
Diagnosis of Fibrous Dysplasia
The initial test recommended in a suspected case fibrous dysplasia is X-ray which shows characteristic mottling and sclerotic changes with a “ground-glass” appearance. Computed Tomography clearly shows cystic and sclerotic lesions with smooth cortical margins and no soft tissue involvement. MRI shows low to isointense lesions on T1 and T2-weighted images, and demonstrates moderate enhancement with gadolinium.
About one-third of patients with fibrous dysplasia present with elevated levels of serum alkaline phosphatase. Fibrous dysplasia should be differentiated from meningioma, Paget’s disease or other osteodystrophies of the skull base, eosinophilic granuloma, Hand-Schuller-Christian disease, and low-grade central osteosarcoma which present in a similar fashion.
Treatment of Fibrous Dysplasia
Fibrous dysplasia has a good prognosis with very little chance of cancer development. The disease is often self-limiting, and usually stabilizes with bone maturation. Treatment is guided by disease severity and progression and could involve simple observation or surgical intervention.
Patients with mild disease are managed with diet and exercise targeted to maintain bone density and regular follow-up with imaging studies. Watch out also for visual impairment due to canal compression.
Bisphosphonates are often prescribed to prevent bone resorption, with improvement in pain, biochemical markers, and resolution of radiographic lesions with regular use.
Surgical intervention is the mainstay of treatment for symptomatic fibrous dysplasia. It is done in case of severe disfigurement, pathologic fractures, and other complications such as visual impairment. Therapeutic optic nerve decompression is done to preserve visual function. Recurrence after radical surgical resection is rare, but disease progression remains unabated with conservative surgery.